

مقدمه
مقاله ای در حوزه یادگیری ماشین در ژورنال Nature Method در آوریل 2018 با عنوان Statistics versus Machine learning چاپ شده است، که به خوبی جنبه های مختلف تفاوت یادگیری ماشین و آمار را بررسی کرده است. آنچه در ادامه می آید، ترجمه این مقاله و اضافاتی است که من به آن اضافه کرده ام تا قابل فهم تر و ساده تر شود.
به طور خلاصه
آمار ویژگیهای جامعه را بر اساس نمونه ای که به طور تصادفی از آن استخراج می گردد، استنتاج می کند (برآورد کرده و یا آزمون فرضی در مورد آنها انجام می دهد)؛ ولی، یادگیری ماشینی، الگوهای پیش بینی قابل تعمیم را پیدا می کند.
در ادامه با جزئیات بیشتر به بررسی این دو موضوع میپردازیم:
استنتاج و پیش بینی دو هدف عمده در مطالعه سیستم های مختلف مثلا سیستم های بیولوژیک هستند. استنتاج “یک مدل ریاضی را از فرآیند تولید داده”، ایجاد می کند تا فرضیه ای را در مورد نحوه رفتار سیستم آزمایش کند. در حالی که پیشبینی “نتایج مشاهدهنشده یا رفتارهای آینده” را پیشگویی می کند ( به عنوان مثال: آیا موش با الگوی بیان ژن معین بیماری دارد یا خیر).
در یک پروژه تحقیقاتی، هم استنتاج و هم پیشبینی ارزشمند هستند. علاوه بر آنکه ما به عنوان محقق مایل هستیم رفتارهای یک سیستم را تبیین کنیم (میخواهیم بدانیم فرآیندهای یک سیستم چگونه کار میکنند)، تمایل داریم پیش بینی کنیم در آینده چه اتفاقی خواهد افتاد. برای مثال، ممکن است بخواهیم استنباط کنیم که کدام فرآیندهای بیولوژیکی با اختلال در تنظیم یک ژن در یک بیماری مرتبط است، و همچنین تشخیص دهیم که آیا یک فرد به این بیماری مبتلا است یا خیر و بهترین درمان را پیشبینی کنیم.
بسیاری از روشهای آماری و یادگیری ماشینی (ML) در اصل ممکن است هم برای پیشبینی و هم برای استنتاج استفاده شوند. با این حال، روش های آماری تمرکز طولانی مدت بر استنتاج دارند که از طریق ایجاد و برازش یک مدل احتمال خاص بر داده ها به دست می آید. [ مثلا، در اکثر روشهای آماری ابتدا فرض نرمال بودن را بررسی می کنیم. می خواهیم بدانیم که مدل احتمال نرمال بر داده های ما برازش (فیت) شده است یا نه.]
مدلهای آماری به ما اجازه می دهند تا یک معیار کمی از اطمینان را محاسبه کنیم که یک رابطه کشف شده یک اثر واقعی (true effect) را توصیف می کند که بعید است ناشی از نویز [یا تصادف] باشد. علاوه بر این، اگر دادههای کافی در دسترس باشد، میتوانیم به صراحت مفروضات را تأیید کنیم (مثلاً برابری واریانس های چند گروه) و در صورت نیاز مدل مشخصشده را اصلاح کنیم.
در مقابل، یادگیری ماشین (ML) با استفاده از الگوریتمهای یادگیری همهمنظوره برای یافتن الگوها در دادههای غالباً غنی و سخت (often rich and unwieldy data)، بر پیشبینی تمرکز میکند. روشهای ML مخصوصاً وقتی با «دادههای گسترده (wide data)» سروکار دارند، که در آن تعداد متغیرهای ورودی از تعداد آزمودنی ها بیشتر است، بر خلاف «دادههای طولانی (long data)» که تعداد آزمودنی ها بیشتر از متغیرهای ورودی است، مفید هستند.
ML حداقل فرضیات را در مورد داده ایجاد می کند. این روشها می توانند حتی زمانی که داده ها بدون طراحی آزمایشی که به دقت کنترل می شوند و در حضور برهم کنش های غیرخطی پیچیده جمع آوری شوند، نیز مؤثر باشند.[در آمار برای جمع آوری داده ها از طرح های آزمایش که در آنها کنترل ها بر متغیرهای مداخله کننده اعمال می شود، استفاده می کنند.] با این حال، علیرغم نتایج متقاعدکننده پیشبینی، فقدان یک مدل صریح میتواند راهحلهای ML را برای ارتباط مستقیم با دانش موجود دشوار کند.
آمار کلاسیک و ML از نظر قابلیت محاسباتی با افزایش تعداد متغیرها در هر موضوع متفاوت است. مدلسازی آماری کلاسیک برای دادههایی با چند ده متغیر ورودی و حجم نمونه طراحی شده است؛ که، امروزه کوچک تا متوسط در نظر گرفته میشوند. در این سناریو، مدل جنبه های مشاهده نشده سیستم را پر می کند. با این حال، با افزایش تعداد متغیرهای ورودی و ارتباط احتمالی بین آنها، مدلی که این روابط را به تصویر میکشد پیچیدهتر میشود. بر این اساس، استنباط های آماری دقیق تر می شوند و مرز بین روشهای آماری و ML مبهم تر می شود.
برای مقایسه آمار سنتی با روشهای ML، از شبیه سازی بیان 40 ژن در دو فنوتیپ (-/+) استفاده خواهیم کرد.
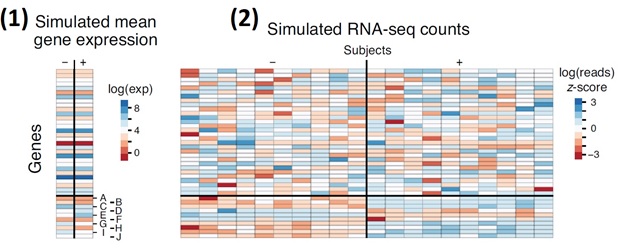
شکل (1) بیان شبیه سازی شده و تعداد خواندن RNA-seq برای 40 ژن که در آن 10 ژن آخر (A-J) به طور متفاوت در دو فنوتیپ بیان شده اند (-/+). (1) میانگین سطوح بیان log شبیهسازی شده برای ژنهای تولید شده با نمونهبرداری از توزیع نرمال با میانگین 4 و انحراف معیار 2. در فنوتیپ + بیان دیفرانسیل ژن های A-J با افزودن یک نرمال استاندارد به هر بیان میانگین در فنوتیپ – ایجاد شد. (2) RNA-seq شبیهسازیشده برای ده آزمودنی در هر فنوتیپ تولید شده از توزیع پواسون بیش از حد بر اساس بیان میانگین در یک تنوع بیولوژیکی، شمارش میکند.

(3) مقادیر P با مقیاس log تنظیم نشده از تجزیه و تحلیل بیان دیفرانسیل آماری به عنوان تابعی از اندازه اثر، که با تغییر برابری در بیان اندازهگیری میشود. (4) مقادیر P با مقیاس ورود به سیستم به عنوان تابعی از اهمیت ژن از طبقهبندی تصادفی جنگل. دایره های قرمز ده ژن متفاوت بیان شده را از شکل 1 مشخص می کنند. (5) توزیع تعداد ژنهای تنظیمنشده به درستی در 1000 شبیهسازی با استنتاج (ناحیه خاکستری) و جنگل تصادفی (خط سیاه) شناسایی شدهاند.
میانگین بیان ژن بین فنوتیپها متفاوت است، اما ما شبیهسازی را طوری تنظیم میکنیم که تفاوت میانگین 30 ژن اول به فنوتیپ مربوط نباشد. ده ژن آخر با تفاوتهای سیستماتیک در میانگین بیان بین فنوتیپها بینظم خواهند بود. برای دستیابی به این هدف، به هر ژن یک بیان log میانگین اختصاص می دهیم که برای هر دو فنوتیپ یکسان است. ژنهای تنظیمنشده (31-40، با برچسب A-J) میانگین بیان آنها در فنوتیپ + مختل شده است (شکل 1). با استفاده از این مقادیر میانگین بیان، ما یک آزمایش RNA-seq را شبیهسازی میکنیم که در آن تعداد مشاهدهشده برای هر ژن از توزیع پواسون با میانگین exp(x + ε) نمونهبرداری میشود، که در آن x میانگین log بیان، منحصر به ژن و فنوتیپ است. و ε ~ N(0, 0.15) به عنوان تنوع زیستی عمل می کند که از سوژه ای به سوژه ای دیگر متفاوت است (شکل 2). برای ژنهای 1-30، که بیان متفاوتی ندارند، z-scores تقریباً N(0, 1) است. برای ژنهای تنظیمنشده، که بیان متفاوتی دارند، z-scores در یک فنوتیپ مثبت است و z-scores در فنوتیپ دیگر منفی است.
هدف ما در شبیه سازی شناسایی ژن هایی است که با فنوتیپ غیر طبیعی مرتبط هستند. ما به طور رسمی این فرضیه صفر را آزمایش خواهیم کرد که میانگین بیان بر اساس فنوتیپ با یک مدل دوجملهای منفی خطی تعمیمیافته (generalized linear negative binomial model) پرکاربرد که امکان تنوع بیولوژیکی را در میان افراد با فنوتیپ یکسان فراهم میکند، متفاوت است. ما یک آزمایش برای هر ژن انجام می دهیم و آنهایی را که تفاوت های آماری معنی داری را در میانگین بیان نشان می دهند، بر اساس مقادیر P تنظیم شده برای آزمایش های چندگانه از طریق روش بنجامینی-هوچبرگ (Benjamini–Hochberg) شناسایی می کنیم. در یک روش بیزی جایگزین، ما احتمال پسین (posterior probability) داشتن بیان دیفرانسیل خاص برای فنوتیپ را محاسبه میکنیم.
شکل 3 مقادیر P آزمایشات بین فنوتیپ ها را به عنوان تابعی از log fold change در بیان ژن نشان می دهد. ده ژن بی نظم با رنگ قرمز مشخص شده اند. استنباط ما 9 مورد از ده مورد (به جز F، با کوچکترین log fold change) را با P <0.05 معنی دار نشان داد. میتوانیم از fold change بهعنوان اندازهگیری اندازه اثر (effect size) با فاصله اطمینان یا بالاترین ناحیه پسین برای نشان دادن عدم قطعیت در تخمین، استفاده کنیم. در یک محیط واقع گرایانه، ژن های شناسایی شده ابتدا توسط تجزیه و تحلیل و سپس به صورت آزمایشی اعتبارسنجی می شوند یا با داده های منابع دیگر مانند شبکه های ژنی پیشنهادی یا حاشیه نویسی مقایسه می شوند.
برای پرسیدن یک سؤال بیولوژیکی مشابه با استفاده از ML، ما معمولاً چندین الگوریتم ارزیابی شده با اعتبارسنجی متقابل روی افراد آزمایشی مستقل، یا روشهای راهاندازی با ارزیابی «خارج از نمونه» را امتحان میکنیم تا یکی را با دقت خوب انتخاب کنیم. بیایید از یک طبقهبندیکننده جنگل تصادفی (RF) (random forest (RF) classifier) استفاده کنیم که به طور همزمان همه ژنها را در نظر میگیرد و درختهای تصمیمگیری چندگانه را برای پیشبینی فنوتیپ بدون فرض یک مدل احتمالی برای تعداد خواندهشده، رشد میدهد. نتیجه این طبقه بندی RF با 100 درخت در شکل 4 نشان داده شده است، جایی که مقادیر P از استنتاج کلاسیک به عنوان تابعی از اهمیت ویژگی (feature) ژن رسم شده است. این امتیاز سهم یک ژن معین را در بهبود متوسط طبقه بندی 5 در یک پارتیشن زمانی که درخت برای انتخاب آن ژن تقسیم می شود، کمیت می کند. بسیاری از الگوریتم های ML دارای معیارهای مشابهی هستند که امکان کمی سازی سهم هر ورودی متغیر در طبقه بندی را فراهم می کند. در شبیهسازی ما، هشت ژن از ده ژن با بیشترین اهمیت، از مجموعههای بینظم بودند. ژنهای D و F که کوچکترین تغییرات چینخوردگی را داشتند، در ده تای بالایی قرار نداشتند (شکل 3).
اگر شبیهسازی را 1000 بار انجام دهیم و تعداد ژنهای بینظمشده را که بهدرستی توسط هر دو روش شناسایی شدهاند، بشماریم (بر اساس رد فرضیه صفر کلاسیک با cutoff مقدار P تعدیلشده یا تعمیم الگوی پیشبینیکننده با RF و رتبهبندی اهمیت ده ویژگی برتر) متوجه می شویم که این دو روش نتایج مشابهی دارند. میانگین تعداد ژن های بی نظم شناسایی شده 7.4/10 برای استنتاج و 7.7/10 برای RF است (شکل 5). هر دو روش دارای میانه 8/10 هستند، اما نمونههای بیشتری از شبیهسازیها را مییابیم که تنها 2 تا 5 ژن نامنظم با استنتاج شناسایی شدهاند. این به این دلیل است که روشی که ما فرآیند انتخاب را طراحی کردهایم برای دو روش متفاوت است: استنتاج با یک cutoff مقدار P تنظیم شده انتخاب میشود تا تعداد ژنها متفاوت باشد، در حالی که در RF ما ده ژن انتخابی برتر را انتخاب میکنیم. ما میتوانستیم یک برش برای امتیاز اهمیت اعمال کنیم، اما نمرات مقیاس هدفی ندارند که بر اساس آن آستانه تعیین شود.
ما از دانش از قبل موجود در مورد داده های RNA-seq برای طراحی یک مدل آماری از فرآیند و استنتاج برای تخمین پارامترهای ناشناخته در مدل از داده ها استفاده کرده ایم. در شبیهسازی ما، مدل رابطه بین میانگین تعداد خواندهها (پارامتر) برای هر ژن برای هر فنوتیپ و تعداد خواندهشده مشاهدهشده برای هر موضوع را محصور میکند. خروجی تجزیه و تحلیل آماری یک آماره آزمایشی برای یک فرضیه خاص و مرزهای اطمینان پارامتر (تغییر برابر میانگین، در این مثال) است. در مثال ما، پارامترهای مدل به صراحت به جنبههای بیان ژن مربوط میشوند – تعداد مولکولهای تولید شده با سرعت معین در یک سلول را میتوان مستقیماً تفسیر کرد.
برای اعمال ML، ما نیازی به دانستن هیچ یک از جزئیات در مورد اندازه گیری RNAseq نداریم. تنها چیزی که مهم است این است که کدام ژن برای تشخیص فنوتیپ بر اساس بیان ژن مفیدتر است. چنین تعمیم زمانی بسیار کمک می کند که ما تعداد زیادی متغیر داشته باشیم، مثلاً در یک آزمایش RNA-seq معمولی که ممکن است صدها تا صدها هزار ویژگی داشته باشد (مثلا رونوشت ها) اما حجم نمونه بسیار کمتری داشته باشد.
اکنون آزمایش پیچیده تری را در نظر بگیرید که در آن هر فرد مشاهدات متعددی را از بافت های مختلف انجام می دهد. حتی اگر فقط یک آزمایش آماری رسمی انجام دهیم که دو فنوتیپ را برای هر بافت مقایسه میکند، مشکل آزمایش چندگانه بسیار پیچیده است. افزایش پیچیدگی دادهها ممکن است استنتاج آماری کلاسیک را کمتر قابل بررسی کند. در عوض میتوانیم از روش ML مانند خوشهبندی ژنها یا بافتها یا هر دو برای استخراج الگوهای اصلی در دادهها، طبقهبندی موضوعات و استنتاج درباره فرآیندهای بیولوژیکی که منجر به فنوتیپ میشوند استفاده کنیم. برای سادهسازی تحلیل، میتوانیم یک کاهش ابعاد مانند میانگینگیری اندازهگیریها در ده آزمودنی با هر فنوتیپ برای هر ژن و هر بافت انجام دهیم.
مرز بین استنتاج آماری و ML موضوع بحث است.
برخی از روشها کاملاً در یک حوزه قرار میگیرند، اما بسیاری از آنها در هر دو مورد استفاده قرار میگیرند. به عنوان مثال، روش bootstrap را می توان برای استنتاج آماری استفاده کرد، اما همچنین به عنوان پایه ای برای روش های مجموعه ای مانند الگوریتم RF عمل می کند. آمار از ما میخواهد که مدلی را انتخاب کنیم که دانش ما از سیستم را در بر بگیرد و ML از ما میخواهد با تکیه بر قابلیتهای تجربی آن، یک الگوریتم پیشبینی را انتخاب کنیم. توجیه یک مدل استنتاج معمولاً به این بستگی دارد که آیا احساس می کنیم به اندازه کافی جوهر سیستم را در بر می گیرد یا خیر. انتخاب الگوریتمهای یادگیری الگو اغلب به معیارهای عملکرد گذشته در سناریوهای مشابه بستگی دارد. استنباط و ML مکمل یکدیگر هستند تا ما را به نتایج معنادار بیولوژیکی راهنمایی کنند.

8 پاسخ